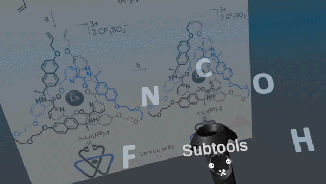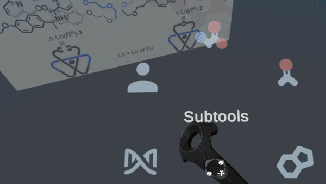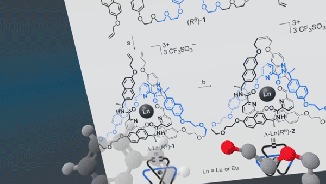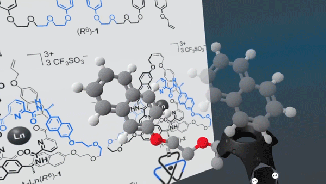
Very excited to announce a beta release of the open-source Narupa VR-enabled builder, which allows the building, designing, and editing of molecules.
Continue reading

Very excited to announce a beta release of the open-source Narupa VR-enabled builder, which allows the building, designing, and editing of molecules.
Continue reading
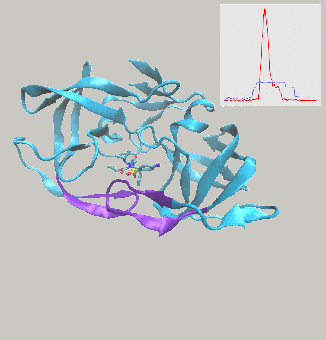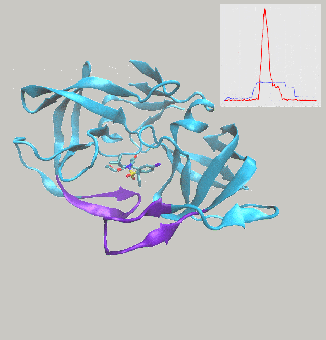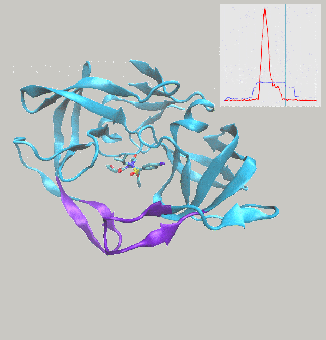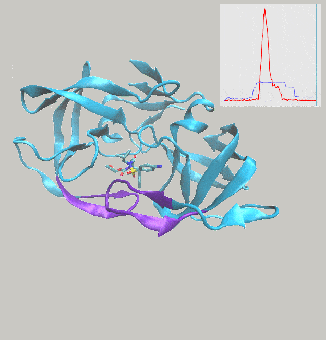
Excited to report on work we’ve just posted to the arXiv, describing interactive molecular dynamics in virtual reality (iMD-VR) using the open-source Narupa framework as an efficient strategy for generating reversible protein-ligand binding and unbinding pathways.
Continue reading

(c) Rachel Freire, Becca Rose, & the Intangible Realities Lab, licensed under CC-BY-SA 4.0
Following on from some preliminary work we published in early 2019, I had an amazing last few days in the Intangible Realities Laboratory, spending time with the fabulous E-textile artists Rachel Freire and Becca Rose making v3 of our open-source VR data gloves, which we have specifically designed to facilitate multi-person interaction of the sort required to carry out careful molecular manipulations during interactive simulations.
Continue reading

Stay tuned, the IRL have just released a prototype for a molecular builder, the latest addition to the open-source Narupa family! Features are being actively developed right now… if you have ideas or features you’d like to see (or bugs to report) let us know via the GitLab issue repository!

New open-access paper from the IRL showing how interactive quantum chemistry in VR can be used to efficiently train Neural Nets to learn potential energy functions. As an ‘Editor’s Choice’ paper, it was featured on the cover, and is already one of the journal’s most read papers.

finally! an open-access paper describing our open-source iMD-VR framework Narupa has been published as in J Chem Phys , and it’s been selected as the “Editor’s Pick featured article”. It’s also scheduled to appear on the journal’s cover, featuring an image made by IRL PhD student Alexander Jamieson-Binnie.

We’ve just published an open access article (arxiv.1902.01827) describing Narupa, our open-source multi-person iMD-VR (interactive molecular dynamics in virtual reality) framework. To go along with the article, we’ve published a stable beta executable of Narupa, available at irl.itch.io/narupaxr.
Continue reading
Over the past few years, we’ve been exploring sound as a sensory channel for understanding the physics of real-time interactive molecular dynamics simulations. In the molecular sciences, sound is a vastly underutilized means for data processing, partly because audio representational standards are less well defined compared to graphics.
Continue reading
New paper posted to the arXiv, which I’m really excited about, where we describe how user-guided real-time interactive quantum mechanics (QM) simulations in virtual reality (iMD-VR) can be used to train neural networks to learn QM energy functions. The paper is entitled “Training neural nets to learn reactive potential energy surfaces using interactive quantum chemistry in virtual reality”.
Continue reading

This is some fun stuff that we’ve been building in the Intangible Realities Laboratory! This video shows my perspective as I tie a knot in 17-Alanine using the Narupa framework while wearing the new customized Etextile VR glove prototype designed by Becca Rose & Rachel Freire.
Continue reading
